
Популярное

7 советов начинающему ип, чтобы избежать проблем (что-то вы точно из этого не знаете)
Недостатки
Как и у любой медали, у этой, под названием ИП тоже есть отрицательные стороны. Самые глобальные...
45
0
0
8 миллиардов не за горами, а что дальше: куда движется население земли и чего ждать всем нам?
Хорошая жизнь
Исследователи отмечают, что для достижения мира, в котором ресурсы одинаково доступны...
67
0
0
9 лайфхаков для владельцев карты «тройка»
Как вернуть деньги с карты Тройка
Карточка «Тройка» в первую очередь предназначена для людей, часто...
47
0
0
12 банков, где выгодно открыть валютный вклад
Сравнительная таблица лучших предложений на сегодня
Сравнение лучших предложений проведем по эффективной...
46
0
0

15 самых выгодных акций российских компаний в 2020 году
Стратегии инвестирования в дивидендные российские акции в 2021 году
Хорошей стратегией станет выбор...
65
0
0

5 самых дерзких ограблений банков в истории
Первый побег
В 1930 году Саттона снова поймали и заключили в тюрьму «Синг-синг». Сидеть 30 лет за решеткой...
45
0
0

4276
Какую информацию в себе несет номер карты
БИН (BIN) – идентификационный номер, установленный банком...
36
0
0
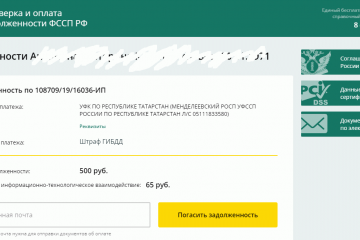
7 разных способов оплатить долг судебным приставам
Что такое Госуслуги?
Портал Госуслуги оснащен единой системой идентификации и аутентификации (ЕСИА),...
43
0
0
Полезные советы

Важно знать!
17 российских брендов одежды, которые ни в чем не уступают зарубежным
SiammSiamm
Бренд, который не боится самовыражения и ярких луков. Он был создан в Екатеринбурге в 2011 выпускницей журфака, Ладой Томиловой. Разрабатывая свой бренд, Лада хотела показать, что каждая девушка...
Читать далее
7 способов получать пассивный доход в 2021 году
4 простых способа быстро связаться с оператором мтс
12 лучших стран по качеству жизни
5 способов сэкономить на покупке валюты
10 идей, как подростку заработать деньги
3 процента на остаток по счету или карте. сколько это и как рассчитать?
14 способов как перечислить деньги на карту сбербанка
10 рублей 1997 года (модификация 2001 года)
1 рубль в украинских гривнах
Рекомендуем






Лучшее

Важно знать!
9 признаков в переписке, что для мужчины вы больше, чем просто знакомая
Кэшбэк для дебетовых карт
Функция кэшбэка доступна для владельцев некоторых стандартных карт, если это предусмотрено условиями банка-эмитента, и для всех премиальных. Многие кредитные учреждения развивают...
Читать далее
10 самых высокооплачиваемых профессий в россии
1 рубль россии: ценные, редкие и дорогие монеты
2b
1 украинская гривна в рублях
50 дней льготного периода в сбербанке по кредитной карте
1000000 вонов в рублях
11 ошибок, которые совершают продавцы на amazon
30 профессий, которые не требуют образования
20 лучших ипотечных программ в москве
Новое








Обсуждаемое

Важно знать!
6 банков, готовых выдать кредит под залог дома
Что делать, если нужна сумма больше, чем дают при рефинансировании
Рефинансирование не позволит вам получить 60-80% от стоимости дорогостоящей недвижимости. Обычно максимальная сумма дополнительных средств,...
Читать далее
10 самых выгодных вкладов в надежных банках в 2021 году
3d-secure защита в сбербанке
10 важных причин отпроситься с работы пораньше
5 лайфхаков по обмену валюты
7 ступень гто 2021
7 видов банковских карт от мастеркард и их отличия
3 простых способа узнать количество миль на карте аэрофлот от сбербанка
10 способов узнать баланс карты «стрелка»
10 крупнейших торговых сетей россии
Популярное







Актуальное
Важно знать!
5 способов связи со службой поддержки сбербанка россии
Контакты Сбербанка
Специалисты технической поддержки для частных клиентов и юридических лиц работают без выходных и перерывов. Связаться со Сбербанком можно с помощью онлайн-инструментов, или позвонив...
Читать далее
5 способов перевести деньги без комиссии
4 способа привязки карты сбербанка к номеру телефону
5 самых необычных банковских карт
7 способов как узнать баланс карты газпромбанк
9 лайфхаков для владельцев карты momentum от сбербанка
4 совета, как разблокировать сим-карту beeline самостоятельно
6 лучших мфо, где можно взять займ онлайн
10 самых крупных заводов россии
8 бесплатных кредитных карт с длинным льготным периодом
Обновления
 5 способов экономить на поездках в московском метро. некоторые вообще ездят бесплатно
5 способов экономить на поездках в московском метро. некоторые вообще ездят бесплатно
Бесплатная пересадка с МЦД на метро по карте «Тройка»
С 20 станций Московских центральных диаметров...
 3 способа перевода средств с билайна на карту
3 способа перевода средств с билайна на карту
Перевод на баланс другого телефона
Если хотите не просто вывести средства с баланса, а собираетесь...
 100 дней без %
100 дней без %
В чем подвох: хитрости и подводные камни
Несмотря на очевидные преимущества кредитной карты «100 дней...
 1 белорусский рубль в рублях
1 белорусский рубль в рублях
Рекомендованные брокеры для работы на финансовых рынках
За 15 лет профессионального трейдинга установился...
 5 рублей 1997 года спмд и ммд
5 рублей 1997 года спмд и ммд
Ценные виды брака и ошибки монетных дворов
Среди нумизматов есть особая категория, коллекционирующих...
 5 типов, что такое финансовая пирамида простыми словами
5 типов, что такое финансовая пирамида простыми словами
Как определить мошенников?
Статья о финансовых пирамидах не может не содержать такой пункт. Стоит сразу...
 16 важных законов, которые изменят жизнь граждан и бизнеса в сентябре 2021 года
16 важных законов, которые изменят жизнь граждан и бизнеса в сентябре 2021 года
С момента наступления какого события закон вступает в силу
Вступлению в силу федеральных законов предшествует...
 Как сменить пароль в сбербанк онлайн: пошаговая инструкция
Как сменить пароль в сбербанк онлайн: пошаговая инструкция
Когда нужно менять пароль от Сбербанк Онлайн
С помощью интернет-банка каждый клиент Сбербанка...
 Дадут ли кредит в тинькофф банке с плохой кредитной историей?
Дадут ли кредит в тинькофф банке с плохой кредитной историей?
Можно ли вернуть кредитную карту
Алгоритм возврата кредитной карты, которой не пользовались, будет также...
 В какие акции вложить деньги в 2021 году: советы и аналитика
В какие акции вложить деньги в 2021 году: советы и аналитика
Риски и способы их избежать
Инвестирование — рискованный бизнес.
На мой взгляд, основные опасности:
Экономический...
 Как найти дешевые авиабилеты?
Как найти дешевые авиабилеты?
Превратитесь в невидимку
Интересный факт: стоимость билета зависит от географического расположения пассажира....
 Президентские выборы в россии (1991)
Президентские выборы в россии (1991)
Трансляция видеонаблюдения за выборами президента
В декабре 2011 года председатель Правительства Российской...
Нашли ошибку, неточность или опечатку в тексте?
Выделите её и нажмите Ctrl + Enter
Выделите её и нажмите Ctrl + Enter